CMP 2.0 employs over 400,000 metagenome-assembled genomes (MAGs), collected from a global collection of more than 30,000 microbiome samples sourced from 9 different human body sites. From this extensive microbiome data collection, we have identified 6,809 bacteria, archaea, and eukaryote species, including many newly discovered species. This is the foundation of our microbiome analysis services.
Our computational pipeline incorporates a proprietary algorithm for detection and abundance estimation of species within the microbiome. Utilizing signature genes that are specific to each species and found across all known strains of that species we provide the most accurate microbiome data available.
When benchmarked against the best competing microbiome profiling methods, CMP 2.0 demonstrates best-in-class performance in terms of accuracy, precision, and sensitivity using both CAMI and NIBSC benchmarks.
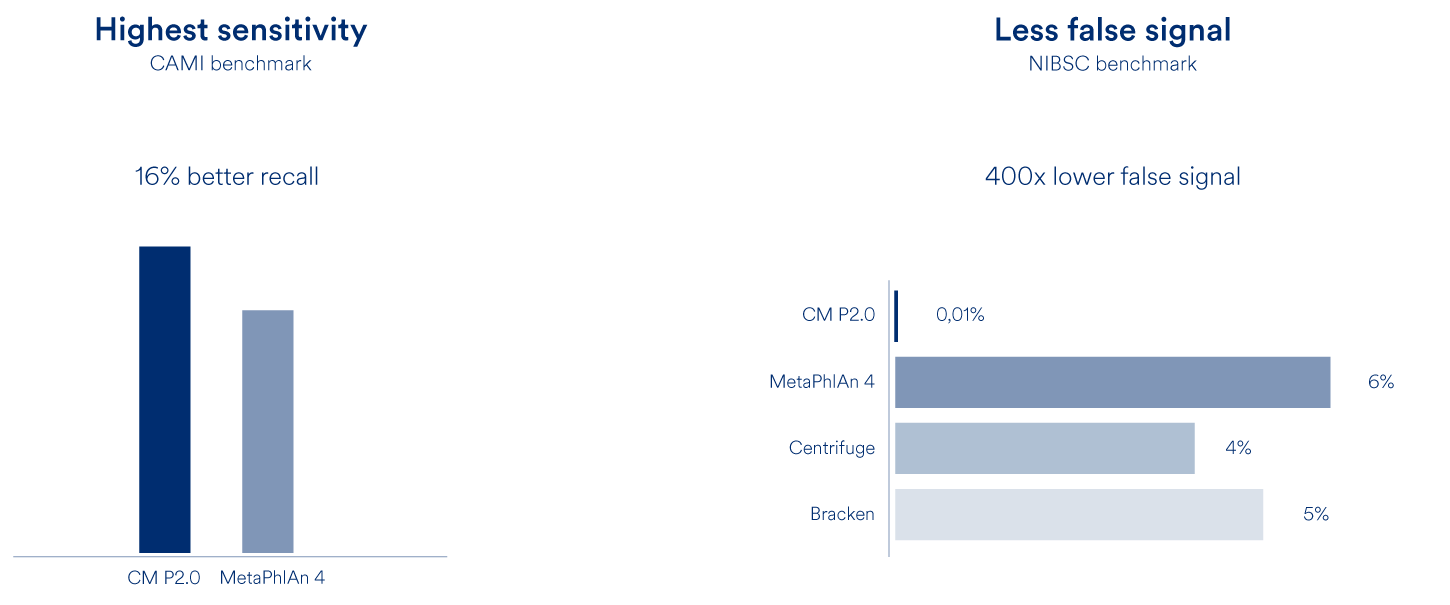
CMP 2.0 provides the most comprehensive, precise and sensitive profiling for the human microbiome. With 6809 species, a recall that is 16% better than competing methods and a false abundance call (FPRA) that is at least 400 times better than any other method CMP 2.0 has no equal.
Furthermore, CMP 2.0 is designed to support microbiome analysis at clonal level, offering superior profiling not just for type strains.
Our approach to strain-level and clonal-level microbiome profiling employs a de novo methodology for detecting and characterizing strains as they appear in the samples. This is crucial since the clonal populations of microbial species are unique to an individual rendering generic reference-based models insufficient for describing the microbiome at this level.
